News
Model Particles in Liquid, Solid, or Gaseous States Using LAMMPS Certified GPU Systems
October 16, 2018
3 min read

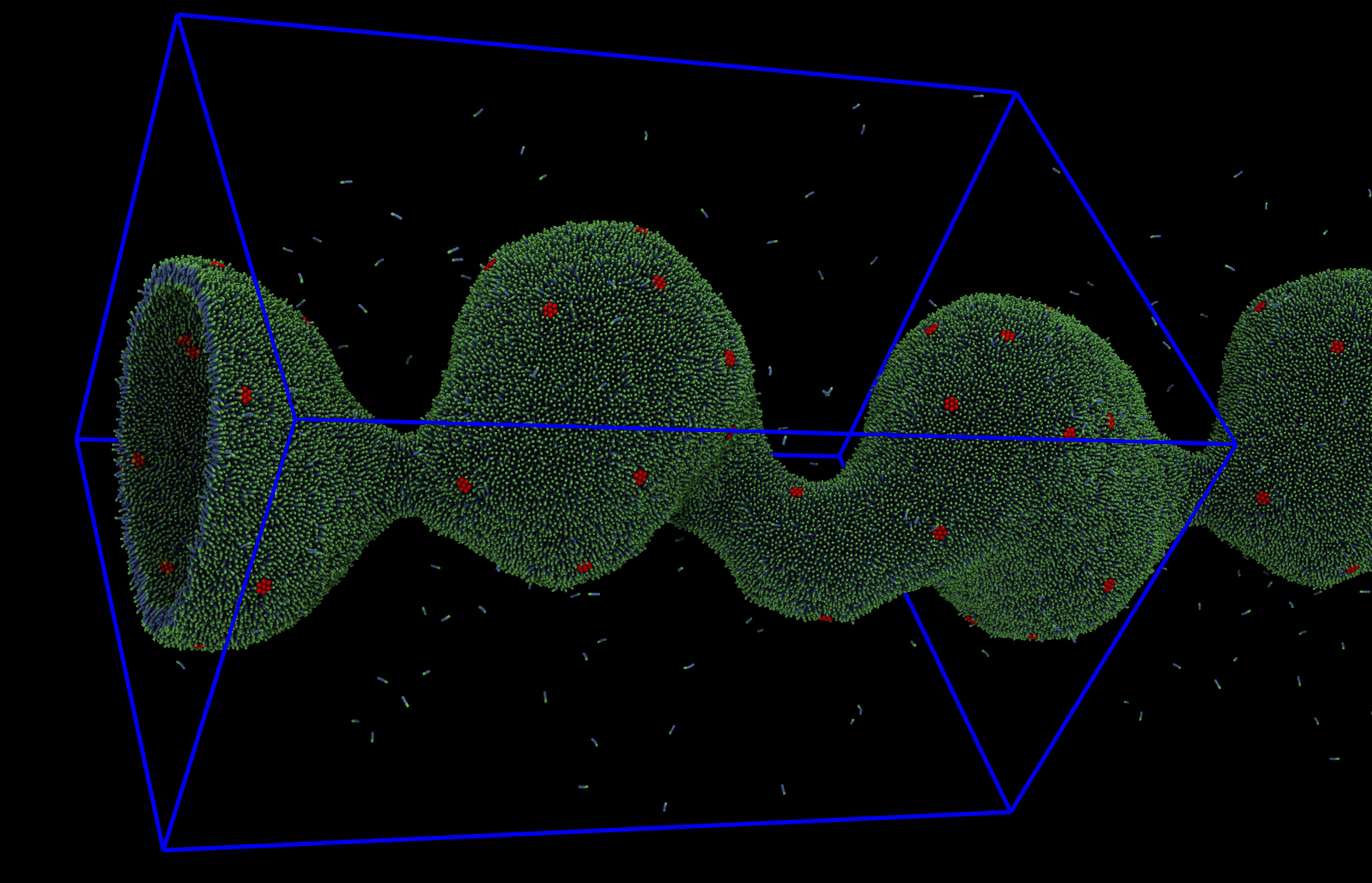
LAMMPS is a classical molecular dynamics (MD) code that models ensembles of particles in a liquid, solid, or gaseous state. It can model atomic, polymeric, biological, solid-state (metals, ceramics, oxides), granular, coarse-grained, or macroscopic systems using a variety of interatomic potentials (force fields) and boundary conditions. It can model 2d or 3d systems with only a few particles up to millions or billions.
LAMMPS can be built and run on a laptop or desktop machine, but is designed for parallel computers. It will run on any parallel machine that supports the MPI message-passing library. This includes shared-memory boxes and distributed-memory clusters and supercomputers.
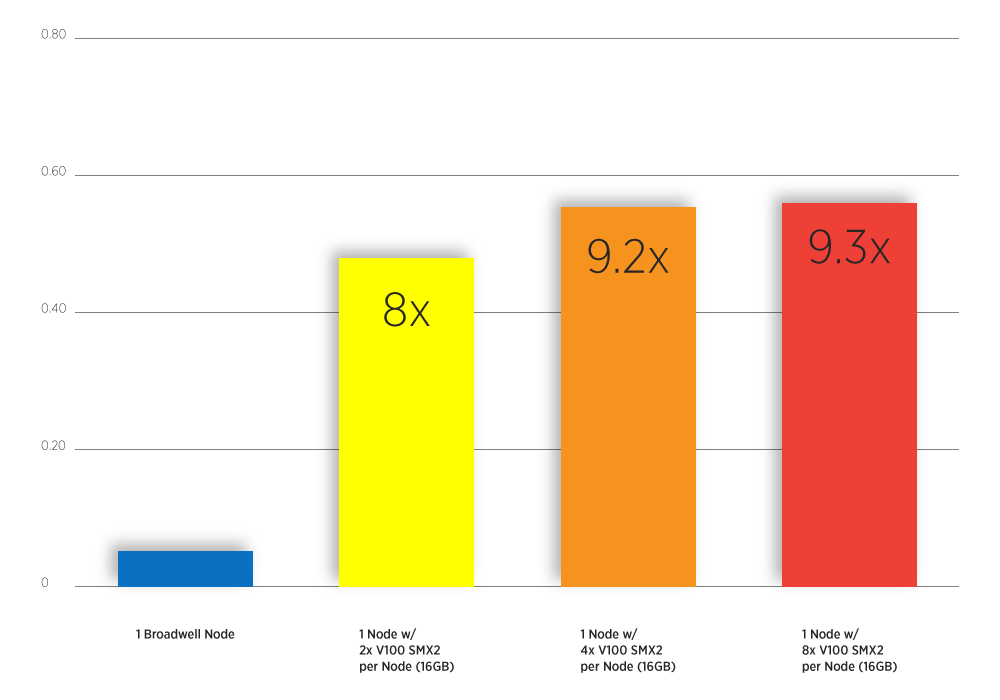
Atomic-Fluid Lennard-Jones 5.0 Cutoff on V100s SMX2. (All nodes contain Dual Intel Xeon E5-2698 v4 2.2GHz CPUs. Untuned on Volta, running LAMMPS v2017.)
LAMMPS is a highly flexible, highly scalable software suite for molecular dyamics developed by Sandia National Laboratories and according to their website: "LAMMPS stands for Large-scale Atomic/Molecular Massively Parallel Simulator. It's a classical molecular dynamics (MD) code. As the name implies, it's designed to run well on parallel machines, but it also runs fine on single-processor desktop machines."
Interested in GPU accelerated systems for LAMMPS? Check out our latest Exxact LAMMPS GPU Systems here!
Or have any questions? Contact us directly here.
LAMMPS is a classical molecular dynamics (MD) code that models ensembles of particles in a liquid, solid, or gaseous state. It can model atomic, polymeric, biological, solid-state (metals, ceramics, oxides), granular, coarse-grained, or macroscopic systems using a variety of interatomic potentials (force fields) and boundary conditions. It can model 2d or 3d systems with only a few particles up to millions or billions.
LAMMPS can be built and run on a laptop or desktop machine, but is designed for parallel computers. It will run on any parallel machine that supports the MPI message-passing library. This includes shared-memory boxes and distributed-memory clusters and supercomputers.
Atomic-Fluid Lennard-Jones 5.0 Cutoff on V100s SMX2. (All nodes contain Dual Intel Xeon E5-2698 v4 2.2GHz CPUs. Untuned on Volta, running LAMMPS v2017.)
LAMMPS is a highly flexible, highly scalable software suite for molecular dyamics developed by Sandia National Laboratories and according to their website: "LAMMPS stands for Large-scale Atomic/Molecular Massively Parallel Simulator. It's a classical molecular dynamics (MD) code. As the name implies, it's designed to run well on parallel machines, but it also runs fine on single-processor desktop machines."
Interested in GPU accelerated systems for LAMMPS? Check out our latest Exxact LAMMPS GPU Systems here!
Or have any questions? Contact us directly here.
