Scientific Computing
How to Install ColabFold & Run AI Protein Folding Locally
December 13, 2023
13 min read


Proteins perform structural, mechanical, chemical, and signaling roles in cells. Designed or discovered proteins are increasingly important; the production of effective pharmaceutical drugs is a lucrative market and also delivers the potential to change the world.
Proteins, boiled down to its simplest for, is a string of amino acids. To solve a protein is to determine the 3D structure based on the amino acid sequence alone. The difficulty of searching and finding the correct structure combinatorically by force is statistically impossible and protein structure prediction has long been considered a futile endeavor. At least it was, until a 60% accurate Alphafold1 demolished other CASP13 competitors, followed by a near 90% accurate AlphaFold2 emerged at CASP14.
Since 2020, progress in structural bioinformatics has continued with academic and open-source alternatives to AlphaFold2. We want to explore AlphaFold’s progress since CASP14 in related models and focus on ColabFold, and how ColabFold cuts protein structure prediction times significantly by improving the decidedly non-glamorous multiple sequence alignment step. The most up to date AlphaFold2 will be addressed as AlphaFold while the first iteration from CASP13 will be addressed as AlphaFold1.
ColabFold is a protein prediction algorithm that is primarily intended to be used in Google’s Colaboratory notebook environment. But, while Google Colab can be OK for small-scale exploratory work, it has limitations for production or serious research without the full unaltered access to high performance accelerators like GPUs and TPUs.
Instead of relying on Google Colab, we’ll go through the step-by-step process of deploying ColabFold locally on your own on-prem machine and demonstrate how to predict the structure of the luciferase enzyme (2d1s in the protein data bank) responsible for the bioluminescence glow of the Japanese firefly.
The development of accurate protein prediction algorithms is deeply rooted in the explosion of harnessing deep learning training to analyze underlying behaviors the human eye is privy to. However, we want to focus on the MSA module that AlphaFold, RoseTTAFold, and OpenFold all use as part of their protein structure prediction recipe.
A multiple sequence alignment (MSA) is, a collection of sequences of similar proteins (or DNA), arranged such that similar stretches of amino acids sequences from different specimen are compared and evaluated. By capturing familiar sequences that share a related structure, the model can access make references to templates to avoid continually predicting a known. Although MSAs extremely useful building a large MSA library of templates can be computationally expensive and may not scale well to large sequence sets.
ColabFold incorporates a fast parallelizable MSA algorithm, called MMSeqs2, to obtain substantial speedups over standard AlphaFold in the total time it takes to predict a sequence. While ColabFold does not stand on its own, it employs either AlphaFold or RoseTTAFold as the structure prediction model and couples it with MMSeqs2.
According to assistant professor Martin Steinegger, co-author of the ColabFold paper who led the development of MMSeqs2, using MMSeqs2 can decrease the time to build MSAs from hours to minutes with the the paper claiming a speedup up of 40 to 60 times.
The key to MSA speedups is a novel strategy of pre-filtering with a k-mer search (a k-mer is a block of amino acids with sequence length 'k'). MMSeqs2 looks for sequences with multiple shared k-mers in a row between sequences. Because MMSeqs2 allows for partial k-mer matches between similar sequence words, it can use longer k-mers and loses only a little sensitivity compared to other MSA search algorithms like jackhmmer, used in the original DeepMind AlphaFold workflow and notebook.
Additionally, because the sequence databases used by MMSeqs2 and the alignments themselves have substantial memory requirements (e.g. about 80 GB of RAM for a sequence of length 342 with about 30 million sequences in the MSA), MMSeqs2 can cache the MSAs for repeated queries, to speed up response times for popular queries. When running the notebook to predict our 2d1s bioluminescence protein, the MMSeqs2 took only 7 seconds to return an MSA versus 60 seconds. Protein engineers that want to keep their sequences private can instead set up their own server to run the GPL 3.0-licensed MMSeqs2.
While ColabFold was developed specifically to be run from Google’s Colaborary cloud notebook environment, its true intent is to make protein folding accessible to all. By delivering a cloud accessible option for protein prediction, democratizing the powerful software enables theorist to experiment. In this section, we’ll go over how to use AlphaFold to predict protein structure, using ColabFold and MMSeqs2 for fast structure predictions.
Cloud notebooks can be great for sharing or presenting work. However, access to GPUs and other hardware can be limited by availability and usage caps.
For more in-depth work, and reproducibility is key it pays to control your environment and all dependencies and why Exxact advocates research institutions opt for an on-prem computing solution. Explore our various high performance multi-GPU computing platforms ranging from workstations to servers to clusters. Configure your ideal system, receive valuable engineer recommendations for your workload, and get a quote today.
In keeping with the goal of making high-quality protein folding more accessible to more people, we will go through the steps to run ColabFold from a local machine. Beyond the accessibility afforded by a cloud notebook, the speedup from incorporating MMSeqs2 for MSAs makes ColabFold worth using on its own. Getting MSAs from MMSeqs2 still involves a server call, but AlphaFold runs locally and its possible to also use ColabFold improvements with RoseTTAFold.
To get started, we’ll initialize and activate a virtual environment to manage dependencies in Python. We use virtualenv, so you may have to modify things a bit if you use Anaconda or miniconda. However, in Python -m venv should work in pretty much the same way. Similarly, we’ll assume a Unix environment, preferably your favorite Linux distribution. If you want to use Windows (e.g. using the Anaconda Unix shell), you may need to make some substantial adjustments to make this work.
To make and activate the virtual environment from the command line:
virtualenv cl_env --python=python3.8
source cl_env/bin/activate
pip install jupyter notebook
Here is a modified version of the ColabFold repository provided by this fork by Rive Sunder, where the notebook has stripped out much of the Colab-specific user-interface, installation commands, and other bits that we won’t use. Otherwise, the original ColabFold repository is linked here by sokrypton (Sergey Ovchinnikov).
# To Use Modified/Stripped ColabFold
git clone git@github.com:rivesunder/ColabFold.git
# To Clone Original and Modify ColabFold on your Own
git clone git@github.com:sokrypton/ColabFold.git
cd ColabFold
When running the notebook in Google colaboratory, there are several cells for installing dependencies. For the modified notebook running locally I’ve stripped the setup code: we only want to run installation once on the command line using a virtual environment (make sure it’s activated!).
The following lines install necessary dependencies and get our environment ready for running the notebook
pip install -q --no-warn-conflicts 'colabfold[alphafold-minus-jax] @ git+https://github.com/sokrypton/ColabFold'
pip install --upgrade dm-haiku
Most likely, the command above installed the base version of Jax without GPU support. jax 0.4.13 and jaxlib 0.4.13 are CPU only versions – To enable GPU support, you will need a more up-to-date version, the more recent the better. You can check with:
#pip freeze | grep jax
Assuming you want to use your local GPU, you can install the right version of jax using the following pattern. CUDA 11 is installed on this machine, but you can check the JAX docs for the proper version for your setup.
# First, uninstall JAX
pip uninstall jax jaxlib
# Next, install JAX with CUDA
pip install --upgrade "jax[cuda11_local]" -f \\ https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
# Verify JAX Version is newer than 0.4.13
pip freeze | grep jax
The next few commands are adapted from the official ColabFold implementation in order to create a symbolic link in the virtual environment to AlphaFold.
ln -s cl_env/lib/python3.8/site-packages/alphafold alphafold
# This next command is not needed for the modified notebook. Otherwise:
#ln -s cl_env/lib/python3.8/site-packages/colabfold colabfold
There’s also a find and replace command to add a clipping function in the AlphaFold file. Make sure the following command is on a single line.
sed -i 's/weights = jax.nn.softmax(logits)/logits=jnp.clip(logits,-1e8,1e8);weights=jax.nn.softmax(logits)/g' alphafold/model/modules.py
Finally, we can write a file that serves as a flag indicating ColabFold is set up and ready to go:
touch COLABFOLD_READY
If everything has been successful, you can now type “jupyter notebook” on the command line to launch a local jupyter server. Navigate to AlphaFold2.ipynb in the ColabFold folder, navigate to Runtime and hit Run All (or Ctrl+F9) run all cells and download AlphaFold parameters and run structure prediction for the Japanese firefly luciferase 2d1s.
If you want to input your own sequence, or mutate the default firefly luciferase, you can modify or replace the sequence assigned to “query_sequence” in the third code cell of the notebook.
The notebook provides:
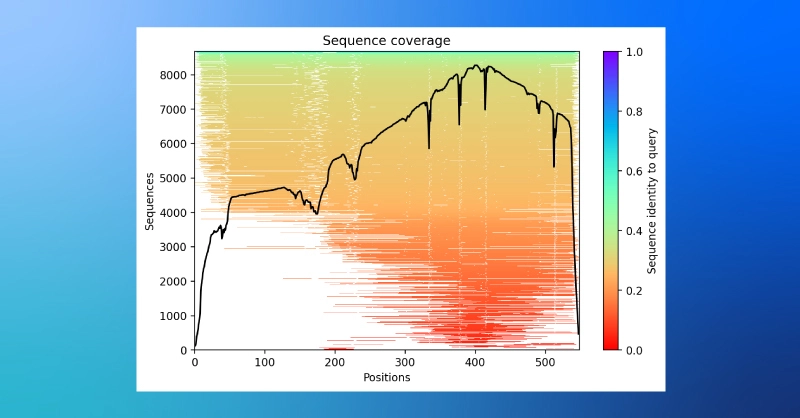
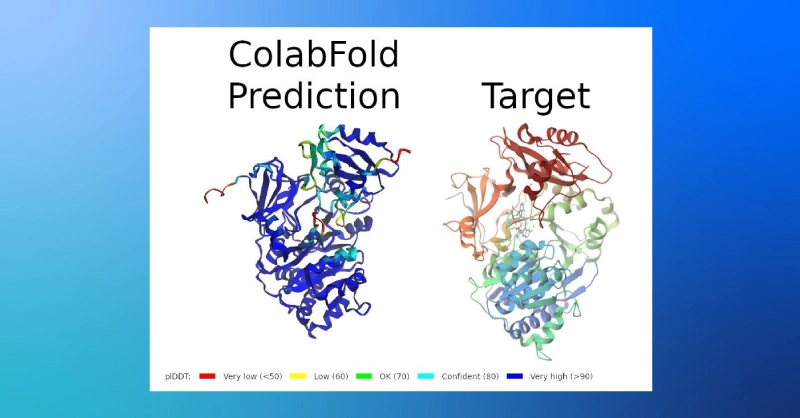
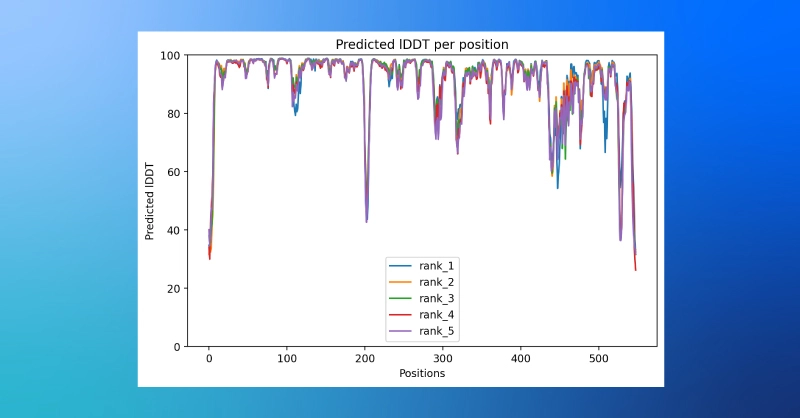
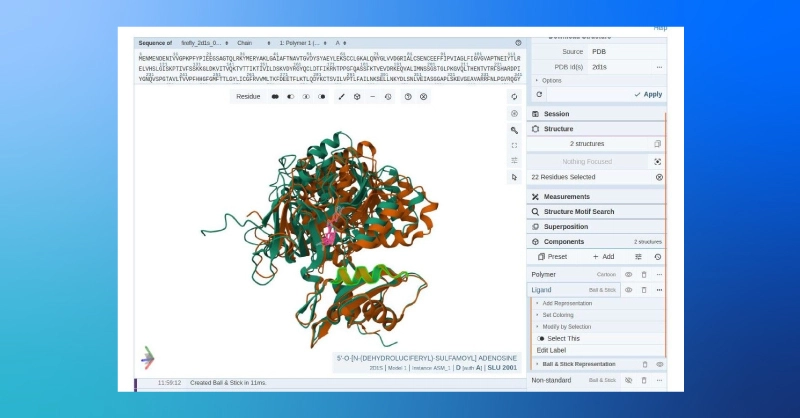
Reading press coverage on the heels of CASP14 in 2020, protein folding enthusiasts can be forgiven for thinking that there’s nothing left to solve in the protein folding problem. In fact, protein structure prediction is just one part of the protein folding problem. Post-translational modification, folding chaperones, and protein glycosylation are a few of the mechanisms that can modify the structure and chemical function of a given protein sequence.
In the years since CASP14, progress has continued. Additional protein structure prediction models have been developed with comparable performance. RoseTTAFold and OpenFold use MSAs, while ESMFold and OmegaFold predict structures from a single sequence without first building an MSA.
An enthusiastic community has arisen, applying hacker mentality to use AlphaFold and peer models for modeling multimers and protein binders, and more. At CASP15, substantial improvements in modeling structures for multimeric proteins and protein-protein interactions, weak-points for the CASP14 AlphaFold. The current released model for AlphaFold released mid 2023 features improved accuracy for docking, nucleic acids, ligand bonding, and more, just another step closer to even more accurate protein prediction. While a 90% confidence is quite accurate, it is not accurate enough for designing a purposeful novel protein with just an amino acid sequence.
The fruits of AlphaFold and subsequent models for the structural biology and protein engineering now include design. The developers of ColabFold have also built ColabDesign, which can use protein structure prediction models to come up with plausible sequences to bind a target or fit a desired structure. Diffusion denoising models have generated substantial interest in image/video generation, and diffusion has recently made strides in the protein structure world as well. RFDiffusion, developed by the Baker Lab and colleagues, as well as other developments in protein structure diffusion models may herald exciting possibilities in bespoke designer proteins for therapeutics, materials sciences, and chemical engineering.
Running AI protein prediction is no easy task for any computer out there, but keeping data close without relying on outside sources for hardware is valuable for data sensitive operations. Exxact delivers high performance workstations, servers, and clusters for molecular discovery all tested, optimized, and validated to the highest standard. Talk to our engineers today to configure your dream machine today.
Proteins perform structural, mechanical, chemical, and signaling roles in cells. Designed or discovered proteins are increasingly important; the production of effective pharmaceutical drugs is a lucrative market and also delivers the potential to change the world.
Proteins, boiled down to its simplest for, is a string of amino acids. To solve a protein is to determine the 3D structure based on the amino acid sequence alone. The difficulty of searching and finding the correct structure combinatorically by force is statistically impossible and protein structure prediction has long been considered a futile endeavor. At least it was, until a 60% accurate Alphafold1 demolished other CASP13 competitors, followed by a near 90% accurate AlphaFold2 emerged at CASP14.
Since 2020, progress in structural bioinformatics has continued with academic and open-source alternatives to AlphaFold2. We want to explore AlphaFold’s progress since CASP14 in related models and focus on ColabFold, and how ColabFold cuts protein structure prediction times significantly by improving the decidedly non-glamorous multiple sequence alignment step. The most up to date AlphaFold2 will be addressed as AlphaFold while the first iteration from CASP13 will be addressed as AlphaFold1.
ColabFold is a protein prediction algorithm that is primarily intended to be used in Google’s Colaboratory notebook environment. But, while Google Colab can be OK for small-scale exploratory work, it has limitations for production or serious research without the full unaltered access to high performance accelerators like GPUs and TPUs.
Instead of relying on Google Colab, we’ll go through the step-by-step process of deploying ColabFold locally on your own on-prem machine and demonstrate how to predict the structure of the luciferase enzyme (2d1s in the protein data bank) responsible for the bioluminescence glow of the Japanese firefly.
The development of accurate protein prediction algorithms is deeply rooted in the explosion of harnessing deep learning training to analyze underlying behaviors the human eye is privy to. However, we want to focus on the MSA module that AlphaFold, RoseTTAFold, and OpenFold all use as part of their protein structure prediction recipe.
A multiple sequence alignment (MSA) is, a collection of sequences of similar proteins (or DNA), arranged such that similar stretches of amino acids sequences from different specimen are compared and evaluated. By capturing familiar sequences that share a related structure, the model can access make references to templates to avoid continually predicting a known. Although MSAs extremely useful building a large MSA library of templates can be computationally expensive and may not scale well to large sequence sets.
ColabFold incorporates a fast parallelizable MSA algorithm, called MMSeqs2, to obtain substantial speedups over standard AlphaFold in the total time it takes to predict a sequence. While ColabFold does not stand on its own, it employs either AlphaFold or RoseTTAFold as the structure prediction model and couples it with MMSeqs2.
According to assistant professor Martin Steinegger, co-author of the ColabFold paper who led the development of MMSeqs2, using MMSeqs2 can decrease the time to build MSAs from hours to minutes with the the paper claiming a speedup up of 40 to 60 times.
The key to MSA speedups is a novel strategy of pre-filtering with a k-mer search (a k-mer is a block of amino acids with sequence length 'k'). MMSeqs2 looks for sequences with multiple shared k-mers in a row between sequences. Because MMSeqs2 allows for partial k-mer matches between similar sequence words, it can use longer k-mers and loses only a little sensitivity compared to other MSA search algorithms like jackhmmer, used in the original DeepMind AlphaFold workflow and notebook.
Additionally, because the sequence databases used by MMSeqs2 and the alignments themselves have substantial memory requirements (e.g. about 80 GB of RAM for a sequence of length 342 with about 30 million sequences in the MSA), MMSeqs2 can cache the MSAs for repeated queries, to speed up response times for popular queries. When running the notebook to predict our 2d1s bioluminescence protein, the MMSeqs2 took only 7 seconds to return an MSA versus 60 seconds. Protein engineers that want to keep their sequences private can instead set up their own server to run the GPL 3.0-licensed MMSeqs2.
While ColabFold was developed specifically to be run from Google’s Colaborary cloud notebook environment, its true intent is to make protein folding accessible to all. By delivering a cloud accessible option for protein prediction, democratizing the powerful software enables theorist to experiment. In this section, we’ll go over how to use AlphaFold to predict protein structure, using ColabFold and MMSeqs2 for fast structure predictions.
Cloud notebooks can be great for sharing or presenting work. However, access to GPUs and other hardware can be limited by availability and usage caps.
For more in-depth work, and reproducibility is key it pays to control your environment and all dependencies and why Exxact advocates research institutions opt for an on-prem computing solution. Explore our various high performance multi-GPU computing platforms ranging from workstations to servers to clusters. Configure your ideal system, receive valuable engineer recommendations for your workload, and get a quote today.
In keeping with the goal of making high-quality protein folding more accessible to more people, we will go through the steps to run ColabFold from a local machine. Beyond the accessibility afforded by a cloud notebook, the speedup from incorporating MMSeqs2 for MSAs makes ColabFold worth using on its own. Getting MSAs from MMSeqs2 still involves a server call, but AlphaFold runs locally and its possible to also use ColabFold improvements with RoseTTAFold.
To get started, we’ll initialize and activate a virtual environment to manage dependencies in Python. We use virtualenv, so you may have to modify things a bit if you use Anaconda or miniconda. However, in Python -m venv should work in pretty much the same way. Similarly, we’ll assume a Unix environment, preferably your favorite Linux distribution. If you want to use Windows (e.g. using the Anaconda Unix shell), you may need to make some substantial adjustments to make this work.
To make and activate the virtual environment from the command line:
virtualenv cl_env --python=python3.8
source cl_env/bin/activate
pip install jupyter notebook
Here is a modified version of the ColabFold repository provided by this fork by Rive Sunder, where the notebook has stripped out much of the Colab-specific user-interface, installation commands, and other bits that we won’t use. Otherwise, the original ColabFold repository is linked here by sokrypton (Sergey Ovchinnikov).
# To Use Modified/Stripped ColabFold
git clone git@github.com:rivesunder/ColabFold.git
# To Clone Original and Modify ColabFold on your Own
git clone git@github.com:sokrypton/ColabFold.git
cd ColabFold
When running the notebook in Google colaboratory, there are several cells for installing dependencies. For the modified notebook running locally I’ve stripped the setup code: we only want to run installation once on the command line using a virtual environment (make sure it’s activated!).
The following lines install necessary dependencies and get our environment ready for running the notebook
pip install -q --no-warn-conflicts 'colabfold[alphafold-minus-jax] @ git+https://github.com/sokrypton/ColabFold'
pip install --upgrade dm-haiku
Most likely, the command above installed the base version of Jax without GPU support. jax 0.4.13 and jaxlib 0.4.13 are CPU only versions – To enable GPU support, you will need a more up-to-date version, the more recent the better. You can check with:
#pip freeze | grep jax
Assuming you want to use your local GPU, you can install the right version of jax using the following pattern. CUDA 11 is installed on this machine, but you can check the JAX docs for the proper version for your setup.
# First, uninstall JAX
pip uninstall jax jaxlib
# Next, install JAX with CUDA
pip install --upgrade "jax[cuda11_local]" -f \\ https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
# Verify JAX Version is newer than 0.4.13
pip freeze | grep jax
The next few commands are adapted from the official ColabFold implementation in order to create a symbolic link in the virtual environment to AlphaFold.
ln -s cl_env/lib/python3.8/site-packages/alphafold alphafold
# This next command is not needed for the modified notebook. Otherwise:
#ln -s cl_env/lib/python3.8/site-packages/colabfold colabfold
There’s also a find and replace command to add a clipping function in the AlphaFold file. Make sure the following command is on a single line.
sed -i 's/weights = jax.nn.softmax(logits)/logits=jnp.clip(logits,-1e8,1e8);weights=jax.nn.softmax(logits)/g' alphafold/model/modules.py
Finally, we can write a file that serves as a flag indicating ColabFold is set up and ready to go:
touch COLABFOLD_READY
If everything has been successful, you can now type “jupyter notebook” on the command line to launch a local jupyter server. Navigate to AlphaFold2.ipynb in the ColabFold folder, navigate to Runtime and hit Run All (or Ctrl+F9) run all cells and download AlphaFold parameters and run structure prediction for the Japanese firefly luciferase 2d1s.
If you want to input your own sequence, or mutate the default firefly luciferase, you can modify or replace the sequence assigned to “query_sequence” in the third code cell of the notebook.
The notebook provides:
Reading press coverage on the heels of CASP14 in 2020, protein folding enthusiasts can be forgiven for thinking that there’s nothing left to solve in the protein folding problem. In fact, protein structure prediction is just one part of the protein folding problem. Post-translational modification, folding chaperones, and protein glycosylation are a few of the mechanisms that can modify the structure and chemical function of a given protein sequence.
In the years since CASP14, progress has continued. Additional protein structure prediction models have been developed with comparable performance. RoseTTAFold and OpenFold use MSAs, while ESMFold and OmegaFold predict structures from a single sequence without first building an MSA.
An enthusiastic community has arisen, applying hacker mentality to use AlphaFold and peer models for modeling multimers and protein binders, and more. At CASP15, substantial improvements in modeling structures for multimeric proteins and protein-protein interactions, weak-points for the CASP14 AlphaFold. The current released model for AlphaFold released mid 2023 features improved accuracy for docking, nucleic acids, ligand bonding, and more, just another step closer to even more accurate protein prediction. While a 90% confidence is quite accurate, it is not accurate enough for designing a purposeful novel protein with just an amino acid sequence.
The fruits of AlphaFold and subsequent models for the structural biology and protein engineering now include design. The developers of ColabFold have also built ColabDesign, which can use protein structure prediction models to come up with plausible sequences to bind a target or fit a desired structure. Diffusion denoising models have generated substantial interest in image/video generation, and diffusion has recently made strides in the protein structure world as well. RFDiffusion, developed by the Baker Lab and colleagues, as well as other developments in protein structure diffusion models may herald exciting possibilities in bespoke designer proteins for therapeutics, materials sciences, and chemical engineering.
Running AI protein prediction is no easy task for any computer out there, but keeping data close without relying on outside sources for hardware is valuable for data sensitive operations. Exxact delivers high performance workstations, servers, and clusters for molecular discovery all tested, optimized, and validated to the highest standard. Talk to our engineers today to configure your dream machine today.
