Scientific Computing
Protein Structure Prediction & Drug Discovery with AlphaFold and RoseTTAFold
April 28, 2023
17 min read


For proteins, structure drives function. Proteins attain their functional forms via an intricate folding process. As one of the most abundant of the four major biological macromolecules in living things, understanding life means understanding proteins.
As much as their structure and function drive the processes of living things, the wrong protein structure in the wrong place causes illness. To mitigate the symptoms, and sometimes cure, a disease means to understand and intervene with the mutated proteins. For many diseases, whether caused by a virus, a bacterial pathogen, or our own aberrant proteins, treating disease means targeting the proteins involved.
Penicillin, the world’s most famous antibiotic, kills bacteria by binding to and inhibiting the protein enzymes used by bacteria to build cell walls made up of peptidoglycans. While penicillin is known to be discovered by accident, modern drug development now has the tools to intentionally design drugs to treat diseases.
Modern deep learning techniques have recently made incredible contributions to the decades-old grand challenge in science: predicting the structure of proteins based solely on the amino acid sequence. By some estimates, the protein structure prediction problem was largely solved by the second iteration of DeepMind’s AlphaFold at the Critical Assessment of Protein Structure Prediction 14 (CASP14) in 2020. AlphaFold makes use of techniques from contemporary natural language processing with attention-based transformer models, as well as conventional bioinformatics methods to accurately predict the structure of a protein, with no other algorithms coming close.
AlphaFold and similar models inspired by its development have led to a rapid rate of progress in structural biology. Not surprisingly, many see the breakthrough as a means to develop treatments for previously un-druggable diseases. In this article, we’ll discuss the background of the protein structure prediction problem and what the introduction of accessible and accurate protein structure estimates means for drug development. We’ll also go over some of what’s been done so far and discuss how to get started working with AlphaFold and RoseTTAFold models, including publicly available online resources and hardware and software considerations.
Every living thing on earth relies on proteins to function. Even viruses, which aren’t technically considered to be alive, are wholly reliant on proteins produced by host cells for their reproduction.
Alongside lipids, nucleic acids, and carbohydrates, proteins are one of the four major biological macromolecules. Proteins fulfill myriad roles, including generating force and movement (such as the muscle protein myosin), structure (such as microtubules making up cellular cytoskeletons), computation (such as proteins regulating action potentials in neural signaling), and as chemical catalysts (any number of protein enzymes).
Considering that they are constructed from a 1-dimensional sequence of amino acids, make proteins extremely fascinating. The folding of a complicated protein complex is like building a jet engine out of a set of strings, using just the right patterns of beads.
Knowledge of protein structures informs how they operate in health and disease and understanding protein structure can reveal insights into modifying disease behavior and developing drugs to tackle the most difficult pathologies.
Protein structure databases have also grown, but evaluating the structure of a given protein via cryo-EM and x-ray crystallography is a challenging process that can take years.
Until recently, computationally predicting an accurate protein structure from sequence wasn’t much easier, and generally wasn’t very accurate. To understand why, it helps to consider the scale of the problem.
Proteins vary in size and the boundary at which a polymer of amino acids goes from being a small peptide to qualifying for classification as a protein. Most proteins are significantly smaller: a few hundred amino acid residues on average but can span to the thousands. The largest known protein is titin, range from being 27,000 to about 35,000 amino acids long.
We could search through all possible conformations of a given amino acid sequence and keep the most favorable, right? Doing so might take more time than the age of the universe. A protein with just 100 amino acid residues would have 3198 or an immeasurable 1059 different structures to consider. Apply that to titin and scientific discovery would be at a standstill.
The potential of AlphaFold to enhance medicine and healthcare was a revolutionary advancement. Although the progress toward personalized medicine and genome sequencing for individuals is still in development, AlphaFold's ability to provide accurate protein structure predictions holds great promise for advancing drug design. There are already many exciting projects underway exploring its numerous applications.
In 2021, about a week after publishing the AlphaFold2 paper, DeepMind announced the AlphaFold Protein Structure Database. The database expanded protein structure coverage of the human proteome (the set of all proteins encoded in the human genome) from about 17% to over 98% at the amino acid residue level.
Powering AI models for ground-breaking discovery requires the best-performing systems. Explore GPU servers powered by AMD EPYC.

A few months later, DeepMind expanded the coverage to include 360,000 protein structure predictions from 21 additional model organisms. The release was coupled with a number of improvements to programmatic access to the database.
The database is publicly available, a significant step toward removing barriers to working with AlphaFold structures and a significant contribution in distributing its tools to the world. The end-to-end pipeline for predicting structures with AlphaFold or RoseTTAFold models comes with significant hardware requirements, energy costs, and run times.
Both models require a high-performance computer with significant storage space, multiple fast CPU cores, plenty of RAM for sequence searching during alignment, and a multi-GPU configuration with ample memory and compute for structure prediction.
The public database reduces wasted energy by reducing the need for different parties to redundantly generate the same structures. It also makes it easier for research labs and pharmaceutical developers to take advantage of the structures even if they don’t have the computational resources to generate structures of their own.
The AlphaFold Protein Database facilitates the development of small molecule therapeutics based on predicted structures but there’s still a need for generating high-quality structure predictions for designing protein therapeutics, and for exploring multiple possible conformational states.
By integrating AlphaFold into a pre-existing target identification and small molecule generation pipeline, Ren et al. demonstrated efficiency improvements in generating ‘hits,’ small molecules capable of binding to a protein involved in a target disease. Hit generation and verification are just the first few steps in a long path to developing a new drug with modern advancements.
Ren and colleagues used a commercially available software package, PandaOmics, to identify cyclin-dependent kinase 20 (CDK20) as a viable protein target for their disease-of-interest, hepatocellular carcinoma (HCC). They then downloaded one of the predicted structures for CDK20 from the AlphaFold Protein Structure Database and used it in conjunction with the commercially available generative chemistry platform Chemistry42 to generate nearly 10,000 small molecules as binding partner candidates.
Various filters for developability whittled that total down to 7 molecules, to assess in the wet lab. Of those 7 candidates one of them was found to have a high binding affinity (Kd = 9.2). They re-iterated the computational chemical generation step based on a proposed mechanism of action for their hit molecule, which yielded a further 24x improvement in binding affinity.
They verified that their best candidate molecule decreased CDK20 kinase activity in a biochemical assay, and also preferentially decreased cellular proliferation in a hepatocellular carcinoma cell line as compared to a non-cancerous cell line, HEK 293.
Ren and her team exhibit AlphaFold's integration in their drug design pipeline, with significant efficiency and speed. The entire process took 30 days, only synthesizing 7 compounds in the first round, plus 6 additional in the second refinement step. What a time to be alive.
One of the ways proteins interact with other proteins and small molecules is by binding. An enzyme often have a binding pockets adjacent to the protein’s active site, in which attached molecules change the enzyme's behavior. By blocking the substrate (binding in the same pocket) or change the shape of the protein (binding elsewhere) called allosteric regulation.
The two main ways of thinking about protein binding are the lock and key model, and the hand and glove model.
In reality, both models are useful, and neither is entirely correct. Cryptic pockets are potential binding sites that don’t appear in experimentally determined protein structures like cryo-EM and x-ray crystallography. Proteins typically have to be purified and crystallized a process that can change the native structure.
Recently, Meller et al. demonstrated the use of AlphaFold to discover cryptic pockets. By sub-sampling a protein’s multiple sequence alignment, they showed they could generate a diverse set of protein structures. These variations on a given protein structure may be more similar to the dynamic and flexible states of proteins in vivo useful as seed structures for molecular dynamics simulations.
They applied their method to 10 proteins known to have cryptic binding pockets and were able to recover the cryptic pockets present in 6 of them. Their dataset included 5 structures that were not deposited in the Protein Data Bank at the time AlphaFold was trained, of which they successfully recovered 3 cryptic binding pockets.
The work of Meller et al. demonstrates the potential of using good quality structure models from AlphaFold to design and discover inhibitors for proteins previously considered to be un-druggable through conventional means. They also showed the potential for complementary application of protein structures from AlphaFold with physics-based molecular dynamics simulations using GROMACS.
In the interim between the CASP14 competition and the release of the AlphaFold2 paper and code, the 3-track network model, RoseTTAFold, was developed by the Baker Lab at the University of Washington.
Due to training strategies adopted to work within hardware memory limitations, the researchers found that RoseTTAFold can directly generate structures for multiple interacting proteins.
The first stage of RoseTTAFold is trained on cropped sequences, features of which are then used by the structure module. As a result, RoseTTAFold can be used to predict complex structures for sequences from separate proteins, skipping the typical docking simulation step after structure generation.
Not to be left behind, DeepMind developed a version of AlphaFold trained specifically on protein complexes, dubbed AlphaFold Multimer. Protein-protein or peptide-protein interactions are another viable strategy for developing therapeutics, and several teams have put together demonstrations of how RoseTTAFold and AlphaFold Multimer can help.
Åkhe and Wallner showed that by perturbing the parameters in AlphaFold Multimer they could expand the model’s ability to explore conformational space. They report improved capabilities of AlphaFold used as an alternative to protein-peptide docking simulations, a noteworthy advantage for designing peptide drugs.
To make these kinds of studies more streamlined, AlphaPullown is a Python package developed by Yu, Kosinski, and colleagues for high-throughput screening of protein-protein interactions using AlphaFold Multimer.
In addition to making effective targets for modifying disease presentation, proteins are a promising substrate for engineered therapeutics. Protein drugs are nothing new, with recombinant insulin (a peptide drug) approved by the FDA in 1982, and the well-known autoimmune disease-modifying fusion protein, etanercept, first approved in 1998. The availability of accurate folding prediction tools like AlphaFold and RoseTTAFold holds substantial promise for the computational design of these types of therapeutics.
The human immune system makes heavy use of antibodies to specifically bind to pathogenic cells or proteins, rendering them inoperable and flagging them for destruction by T-cells and macrophages.
In the past few decades, exogenous antibodies have been used to combat difficult diseases like autoimmune disorders, cancers, and even the SARS-CoV-2 virus. One of the earliest monoclonal antibodies to be approved by the FDA, adalimumab (brand name Humira), was the top-selling drug of all time until recently.
Monoclonal antibodies have subsequently experienced substantial research, development, and investment interest, with 100s of approved antibody drugs available and more considered for regulatory approval each year.
Evolution has selected a convenient, partitioned structure in antibodies. The highly variable complementarity determining region (CDR) provides target specificity in the fragment of antigen-binding (Fab), while the crystallization fragment (Fc) provides stability. This means that protein engineers can focus on a relatively short sequence of amino acids when designing a new therapeutic antibody.
The intrinsic design advantages of antibodies come with a few challenges of their own. Namely, AlphaFold performs somewhat worse in predicting the structure of the loops in the CDR, essential for therapeutic efficacy. Specialized models for antibody variable region structures include ABLooper, developed in Charlotte Dean’s lab at Oxford University, as well as IgFold and DeepAb, both developed by Jeffrey Ruffolo, Jeffrey Gray, and colleagues at Johns Hopkins University were inspired by AlphaFold. Even its shortcomings enable researchers to adopt AlphaFold's training ideologies to develop better tools to solve their specific focus.
The rapid pace of development in protein structure prediction since CASP13 and CASP14 provide the tools for significant contributions to improving human health and understanding and mitigating disease. A number of efforts highlight the usefulness of AI and ML protein structure prediction for developing new therapeutics, from small molecules to peptides and protein drugs. The next few years promise to be a productive time for treating difficult pathologies, including the development of treatments for “un-druggable” protein targets.
The hardware and software requirements for the effective application of AlphaFold and company bears some similarity to deep learning as well as more conventional bioinformatics pipelines. Made up of deep learning (neural) building blocks, AlphaFold uses a two-stage model consisting of the “evoformer” and structure models. The neural components of RoseTTAFold make heavy use of rotation and translation invariant SE(3) transformer modules in its 3-track design, incorporating 1D, 2D, and 3D representations.
Both RoseTTAFold and AlphaFold perform best on modern GPUs with a lot of video memory such as the RTX 4090 (24GB) or RTX 6000 Ada (48GB) for inference. In addition to deep learning transformers that run well on GPUs, both methods make use of multiple sequence alignment. Building the multiple sequence alignment for a given protein requires a fast search of large databases, and searching, clustering, and aligning sequences can take up the majority of computation time when predicting structures.
There are a number of different ways in which to use protein folding models or access their output, many of which are free and publicly available. For developing small molecule drugs for protein targets that previously had no experimental structures available in the PDB, you may be able to get everything you need from the AlphaFold Protein Database.
If, on the other hand, a project needs to generate multiple structural variants to explore the possible conformational space for a given protein, (for example to discover druggable cryptic pockets), you would need an implementation with flexibility and control in order to take advantage of the full potential of advanced methods. Users that need more control to explore conformational space with multiple structure variants or design novel sequences for protein or peptide therapeutics may need full control of their own hardware and software.
For turn-key systems with support for AlphaFold and RoseTTAFold models, complemented by reliable support for high-performance molecular dynamics, consider the longstanding expertise with life science workflows from Exxact. We offer workstations, servers, and cluster solutions customized and optimized to your protein structure engineering needs while fulfilling any other Scientific Computing requirements.
For proteins, structure drives function. Proteins attain their functional forms via an intricate folding process. As one of the most abundant of the four major biological macromolecules in living things, understanding life means understanding proteins.
As much as their structure and function drive the processes of living things, the wrong protein structure in the wrong place causes illness. To mitigate the symptoms, and sometimes cure, a disease means to understand and intervene with the mutated proteins. For many diseases, whether caused by a virus, a bacterial pathogen, or our own aberrant proteins, treating disease means targeting the proteins involved.
Penicillin, the world’s most famous antibiotic, kills bacteria by binding to and inhibiting the protein enzymes used by bacteria to build cell walls made up of peptidoglycans. While penicillin is known to be discovered by accident, modern drug development now has the tools to intentionally design drugs to treat diseases.
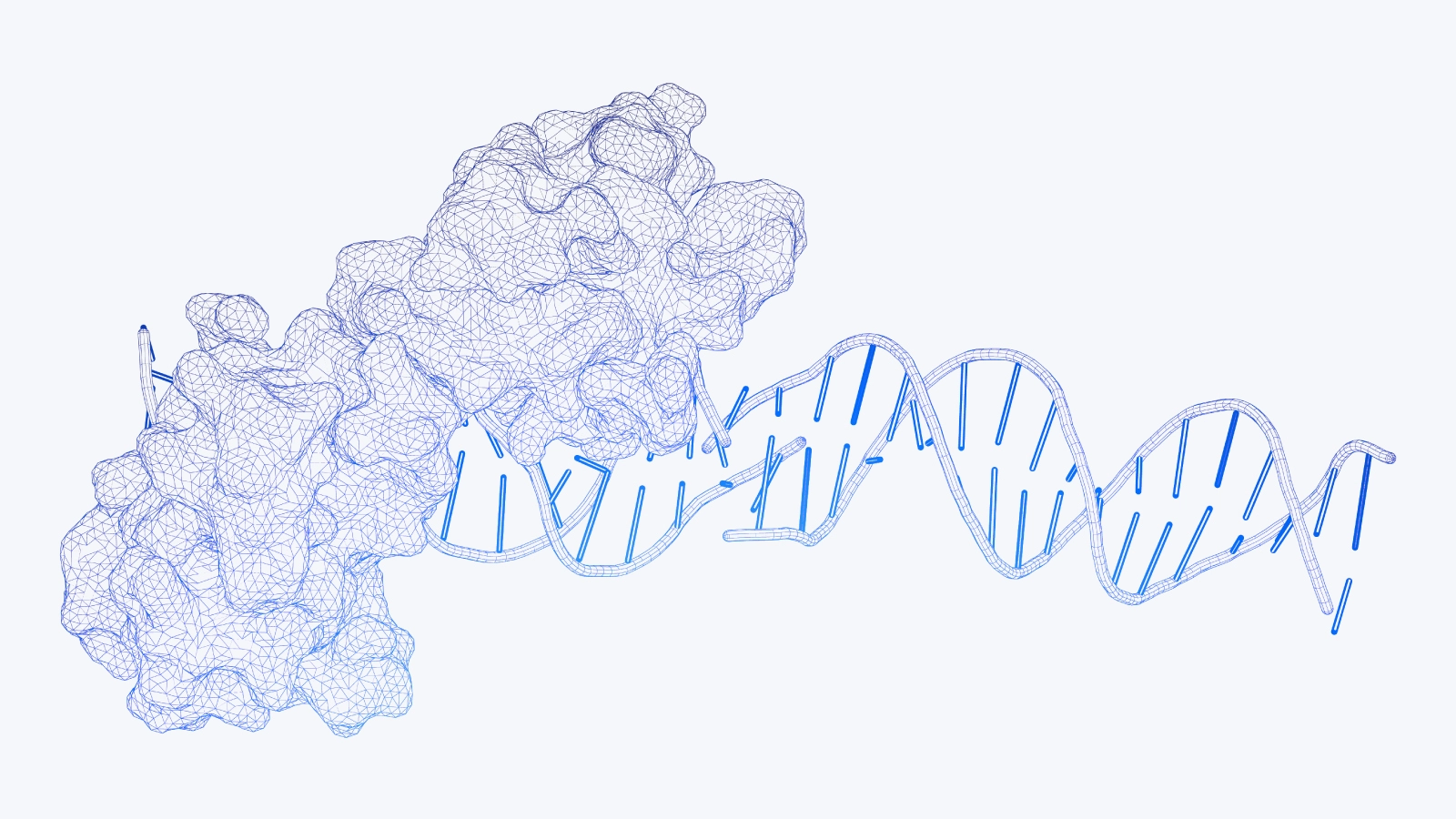
Modern deep learning techniques have recently made incredible contributions to the decades-old grand challenge in science: predicting the structure of proteins based solely on the amino acid sequence. By some estimates, the protein structure prediction problem was largely solved by the second iteration of DeepMind’s AlphaFold at the Critical Assessment of Protein Structure Prediction 14 (CASP14) in 2020. AlphaFold makes use of techniques from contemporary natural language processing with attention-based transformer models, as well as conventional bioinformatics methods to accurately predict the structure of a protein, with no other algorithms coming close.
AlphaFold and similar models inspired by its development have led to a rapid rate of progress in structural biology. Not surprisingly, many see the breakthrough as a means to develop treatments for previously un-druggable diseases. In this article, we’ll discuss the background of the protein structure prediction problem and what the introduction of accessible and accurate protein structure estimates means for drug development. We’ll also go over some of what’s been done so far and discuss how to get started working with AlphaFold and RoseTTAFold models, including publicly available online resources and hardware and software considerations.
Every living thing on earth relies on proteins to function. Even viruses, which aren’t technically considered to be alive, are wholly reliant on proteins produced by host cells for their reproduction.
Alongside lipids, nucleic acids, and carbohydrates, proteins are one of the four major biological macromolecules. Proteins fulfill myriad roles, including generating force and movement (such as the muscle protein myosin), structure (such as microtubules making up cellular cytoskeletons), computation (such as proteins regulating action potentials in neural signaling), and as chemical catalysts (any number of protein enzymes).
Considering that they are constructed from a 1-dimensional sequence of amino acids, make proteins extremely fascinating. The folding of a complicated protein complex is like building a jet engine out of a set of strings, using just the right patterns of beads.

Knowledge of protein structures informs how they operate in health and disease and understanding protein structure can reveal insights into modifying disease behavior and developing drugs to tackle the most difficult pathologies.
Protein structure databases have also grown, but evaluating the structure of a given protein via cryo-EM and x-ray crystallography is a challenging process that can take years.
Until recently, computationally predicting an accurate protein structure from sequence wasn’t much easier, and generally wasn’t very accurate. To understand why, it helps to consider the scale of the problem.
Proteins vary in size and the boundary at which a polymer of amino acids goes from being a small peptide to qualifying for classification as a protein. Most proteins are significantly smaller: a few hundred amino acid residues on average but can span to the thousands. The largest known protein is titin, range from being 27,000 to about 35,000 amino acids long.
We could search through all possible conformations of a given amino acid sequence and keep the most favorable, right? Doing so might take more time than the age of the universe. A protein with just 100 amino acid residues would have 3198 or an immeasurable 1059 different structures to consider. Apply that to titin and scientific discovery would be at a standstill.
The potential of AlphaFold to enhance medicine and healthcare was a revolutionary advancement. Although the progress toward personalized medicine and genome sequencing for individuals is still in development, AlphaFold's ability to provide accurate protein structure predictions holds great promise for advancing drug design. There are already many exciting projects underway exploring its numerous applications.
In 2021, about a week after publishing the AlphaFold2 paper, DeepMind announced the AlphaFold Protein Structure Database. The database expanded protein structure coverage of the human proteome (the set of all proteins encoded in the human genome) from about 17% to over 98% at the amino acid residue level.
Powering AI models for ground-breaking discovery requires the best-performing systems. Explore GPU servers powered by AMD EPYC.
A few months later, DeepMind expanded the coverage to include 360,000 protein structure predictions from 21 additional model organisms. The release was coupled with a number of improvements to programmatic access to the database.
The database is publicly available, a significant step toward removing barriers to working with AlphaFold structures and a significant contribution in distributing its tools to the world. The end-to-end pipeline for predicting structures with AlphaFold or RoseTTAFold models comes with significant hardware requirements, energy costs, and run times.
Both models require a high-performance computer with significant storage space, multiple fast CPU cores, plenty of RAM for sequence searching during alignment, and a multi-GPU configuration with ample memory and compute for structure prediction.
The public database reduces wasted energy by reducing the need for different parties to redundantly generate the same structures. It also makes it easier for research labs and pharmaceutical developers to take advantage of the structures even if they don’t have the computational resources to generate structures of their own.
The AlphaFold Protein Database facilitates the development of small molecule therapeutics based on predicted structures but there’s still a need for generating high-quality structure predictions for designing protein therapeutics, and for exploring multiple possible conformational states.
By integrating AlphaFold into a pre-existing target identification and small molecule generation pipeline, Ren et al. demonstrated efficiency improvements in generating ‘hits,’ small molecules capable of binding to a protein involved in a target disease. Hit generation and verification are just the first few steps in a long path to developing a new drug with modern advancements.
Ren and colleagues used a commercially available software package, PandaOmics, to identify cyclin-dependent kinase 20 (CDK20) as a viable protein target for their disease-of-interest, hepatocellular carcinoma (HCC). They then downloaded one of the predicted structures for CDK20 from the AlphaFold Protein Structure Database and used it in conjunction with the commercially available generative chemistry platform Chemistry42 to generate nearly 10,000 small molecules as binding partner candidates.
Various filters for developability whittled that total down to 7 molecules, to assess in the wet lab. Of those 7 candidates one of them was found to have a high binding affinity (Kd = 9.2). They re-iterated the computational chemical generation step based on a proposed mechanism of action for their hit molecule, which yielded a further 24x improvement in binding affinity.
They verified that their best candidate molecule decreased CDK20 kinase activity in a biochemical assay, and also preferentially decreased cellular proliferation in a hepatocellular carcinoma cell line as compared to a non-cancerous cell line, HEK 293.
Ren and her team exhibit AlphaFold's integration in their drug design pipeline, with significant efficiency and speed. The entire process took 30 days, only synthesizing 7 compounds in the first round, plus 6 additional in the second refinement step. What a time to be alive.
One of the ways proteins interact with other proteins and small molecules is by binding. An enzyme often have a binding pockets adjacent to the protein’s active site, in which attached molecules change the enzyme's behavior. By blocking the substrate (binding in the same pocket) or change the shape of the protein (binding elsewhere) called allosteric regulation.
The two main ways of thinking about protein binding are the lock and key model, and the hand and glove model.
In reality, both models are useful, and neither is entirely correct. Cryptic pockets are potential binding sites that don’t appear in experimentally determined protein structures like cryo-EM and x-ray crystallography. Proteins typically have to be purified and crystallized a process that can change the native structure.
Recently, Meller et al. demonstrated the use of AlphaFold to discover cryptic pockets. By sub-sampling a protein’s multiple sequence alignment, they showed they could generate a diverse set of protein structures. These variations on a given protein structure may be more similar to the dynamic and flexible states of proteins in vivo useful as seed structures for molecular dynamics simulations.
They applied their method to 10 proteins known to have cryptic binding pockets and were able to recover the cryptic pockets present in 6 of them. Their dataset included 5 structures that were not deposited in the Protein Data Bank at the time AlphaFold was trained, of which they successfully recovered 3 cryptic binding pockets.
The work of Meller et al. demonstrates the potential of using good quality structure models from AlphaFold to design and discover inhibitors for proteins previously considered to be un-druggable through conventional means. They also showed the potential for complementary application of protein structures from AlphaFold with physics-based molecular dynamics simulations using GROMACS.
In the interim between the CASP14 competition and the release of the AlphaFold2 paper and code, the 3-track network model, RoseTTAFold, was developed by the Baker Lab at the University of Washington.
Due to training strategies adopted to work within hardware memory limitations, the researchers found that RoseTTAFold can directly generate structures for multiple interacting proteins.
The first stage of RoseTTAFold is trained on cropped sequences, features of which are then used by the structure module. As a result, RoseTTAFold can be used to predict complex structures for sequences from separate proteins, skipping the typical docking simulation step after structure generation.
Not to be left behind, DeepMind developed a version of AlphaFold trained specifically on protein complexes, dubbed AlphaFold Multimer. Protein-protein or peptide-protein interactions are another viable strategy for developing therapeutics, and several teams have put together demonstrations of how RoseTTAFold and AlphaFold Multimer can help.
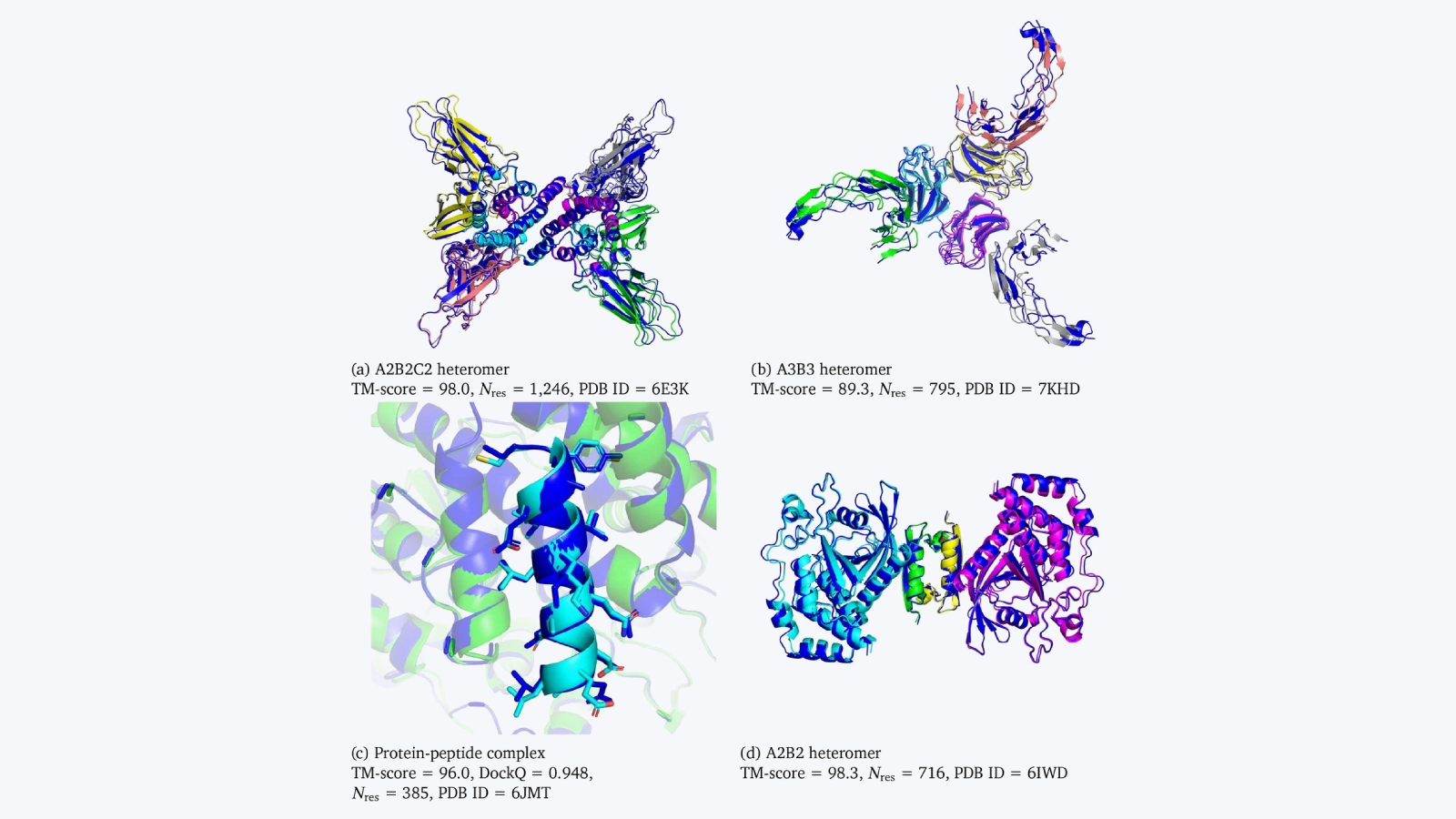
Åkhe and Wallner showed that by perturbing the parameters in AlphaFold Multimer they could expand the model’s ability to explore conformational space. They report improved capabilities of AlphaFold used as an alternative to protein-peptide docking simulations, a noteworthy advantage for designing peptide drugs.
To make these kinds of studies more streamlined, AlphaPullown is a Python package developed by Yu, Kosinski, and colleagues for high-throughput screening of protein-protein interactions using AlphaFold Multimer.
In addition to making effective targets for modifying disease presentation, proteins are a promising substrate for engineered therapeutics. Protein drugs are nothing new, with recombinant insulin (a peptide drug) approved by the FDA in 1982, and the well-known autoimmune disease-modifying fusion protein, etanercept, first approved in 1998. The availability of accurate folding prediction tools like AlphaFold and RoseTTAFold holds substantial promise for the computational design of these types of therapeutics.
The human immune system makes heavy use of antibodies to specifically bind to pathogenic cells or proteins, rendering them inoperable and flagging them for destruction by T-cells and macrophages.
In the past few decades, exogenous antibodies have been used to combat difficult diseases like autoimmune disorders, cancers, and even the SARS-CoV-2 virus. One of the earliest monoclonal antibodies to be approved by the FDA, adalimumab (brand name Humira), was the top-selling drug of all time until recently.
Monoclonal antibodies have subsequently experienced substantial research, development, and investment interest, with 100s of approved antibody drugs available and more considered for regulatory approval each year.
Evolution has selected a convenient, partitioned structure in antibodies. The highly variable complementarity determining region (CDR) provides target specificity in the fragment of antigen-binding (Fab), while the crystallization fragment (Fc) provides stability. This means that protein engineers can focus on a relatively short sequence of amino acids when designing a new therapeutic antibody.
The intrinsic design advantages of antibodies come with a few challenges of their own. Namely, AlphaFold performs somewhat worse in predicting the structure of the loops in the CDR, essential for therapeutic efficacy. Specialized models for antibody variable region structures include ABLooper, developed in Charlotte Dean’s lab at Oxford University, as well as IgFold and DeepAb, both developed by Jeffrey Ruffolo, Jeffrey Gray, and colleagues at Johns Hopkins University were inspired by AlphaFold. Even its shortcomings enable researchers to adopt AlphaFold's training ideologies to develop better tools to solve their specific focus.
The rapid pace of development in protein structure prediction since CASP13 and CASP14 provide the tools for significant contributions to improving human health and understanding and mitigating disease. A number of efforts highlight the usefulness of AI and ML protein structure prediction for developing new therapeutics, from small molecules to peptides and protein drugs. The next few years promise to be a productive time for treating difficult pathologies, including the development of treatments for “un-druggable” protein targets.
The hardware and software requirements for the effective application of AlphaFold and company bears some similarity to deep learning as well as more conventional bioinformatics pipelines. Made up of deep learning (neural) building blocks, AlphaFold uses a two-stage model consisting of the “evoformer” and structure models. The neural components of RoseTTAFold make heavy use of rotation and translation invariant SE(3) transformer modules in its 3-track design, incorporating 1D, 2D, and 3D representations.
Both RoseTTAFold and AlphaFold perform best on modern GPUs with a lot of video memory such as the RTX 4090 (24GB) or RTX 6000 Ada (48GB) for inference. In addition to deep learning transformers that run well on GPUs, both methods make use of multiple sequence alignment. Building the multiple sequence alignment for a given protein requires a fast search of large databases, and searching, clustering, and aligning sequences can take up the majority of computation time when predicting structures.
There are a number of different ways in which to use protein folding models or access their output, many of which are free and publicly available. For developing small molecule drugs for protein targets that previously had no experimental structures available in the PDB, you may be able to get everything you need from the AlphaFold Protein Database.
If, on the other hand, a project needs to generate multiple structural variants to explore the possible conformational space for a given protein, (for example to discover druggable cryptic pockets), you would need an implementation with flexibility and control in order to take advantage of the full potential of advanced methods. Users that need more control to explore conformational space with multiple structure variants or design novel sequences for protein or peptide therapeutics may need full control of their own hardware and software.
For turn-key systems with support for AlphaFold and RoseTTAFold models, complemented by reliable support for high-performance molecular dynamics, consider the longstanding expertise with life science workflows from Exxact. We offer workstations, servers, and cluster solutions customized and optimized to your protein structure engineering needs while fulfilling any other Scientific Computing requirements.
