Scientific Computing
OpenMM: An Open Source Customizable Molecular Simulation Toolkit
August 3, 2023
8 min read


OpenMM is a high-performance molecular dynamics simulation toolkit that allows researchers to study the behavior of molecules and materials at the atomic level. It is an open-source GPU-accelerated biomolecular modeling toolkit for simulation that can be used as an application, library, or flexible programming environment.
OpenMM is open-source supported by a large community of users and developers contributing to its ongoing development and improvement. Its capabilities rival other popular Molecular Dynamics simulation suites and libraries and offers extensive programmability options and a detailed API.
Like other molecular dynamics suites, OpenMM is used to simulate a wide range of molecular systems to gain insights into their behavior and properties to design new materials, develop novel drugs, and gain a more comprehensive understanding of molecular processes.
The key defining attribute of OpenMM is its underlying architecture. OpenMM's foundation is built on C++ (with CUDA and OpenCL capabilities) for high-level computations with a Python-based application layer. Due to its easy syntax and learnability, a Python-based outward-facing interface makes scripting and running simulations extremely simple for those with little programming experience.
As a leading supplier of custom scientific workstations and servers and clusters, Exxact Corporation offers custom configurable OpenMM optimized solutions to accelerate your workflow. Talk to us today for more information!
Being open-source enables OpenMM to address many different purposes and various audiences. Chemists and biologists can utilize and tinker with the Python Application layer to run simulations through OpenMM’s simple UI. They can upload PDB files or cross-platform AMBER or GROMACS files as well. Users unfamiliar with Python can use OpenMM Setup, their Python Script Writer, making it easy to get started experimenting before having to master the more complex aspects of OpenMM.
Application developers can use the computational library to add simulation features to their programs. Since OpenMM is technically an API and library of various functions accessed through Python scripting, developers can access code within OpenMM as functions in their programs. This is especially useful when implementing molecular dynamics inside comprehensive suites: e.g. GROMACS can interface with OpenMM calculations and ACEMD now uses OpenMM as the underlying dynamics engine.
Lastly, experienced molecular dynamics engineers can develop custom algorithms with their formulas and code within OpenMM to develop new methods for novel testing. Customization is a huge plus for developing new simulation algorithms.
OpenMM’s flexibility as a toolkit, application, and library lends to its customizability. Its open-source nature enables those familiar with Python to alter the code to match their research. This also allows for ongoing development and recommendation from peers around the world. There’s a good chance that questions you may have regarding OpenMM can be answered by their forum and feedback is constantly provided to further improve its capabilities.
Developers can also utilize what OpenMM has already contributed to the Molecular Dynamics space for leveraging the expansive library to their own applications and software suites for more niche simulations. Explore their Documentation for more information.
OpenMM’s simulations can be run through a Python script with no more than 20 lines of code. They have plenty of examples of how to use OpenMM right out of the box: These scripts can look as simple as:
from openmm.app import *
from openmm import *
from openmm.unit import *
from sys import stdout
pdb = PDBFile('input.pdb')
forcefield = ForceField('amber14-all.xml', 'amber14/tip3pfb.xml')
system = forcefield.createSystem(pdb.topology, nonbondedMethod=PME,
nonbondedCutoff=1*nanometer, constraints=HBonds)
integrator = LangevinMiddleIntegrator(300*kelvin, 1/picosecond, 0.004*picoseconds)
simulation = Simulation(pdb.topology, system, integrator)
simulation.context.setPositions(pdb.positions)
simulation.minimizeEnergy()
simulation.reporters.append(PDBReporter('output.pdb', 1000))
simulation.reporters.append(StateDataReporter(stdout, 1000, step=True,
potentialEnergy=True, temperature=True))
simulation.step(10000)
But even that might seem daunting for those who haven’t touched Python. OpenMM acknowledges this and supplies a user-friendly interface with a builder that helps create fast simulations with ease.
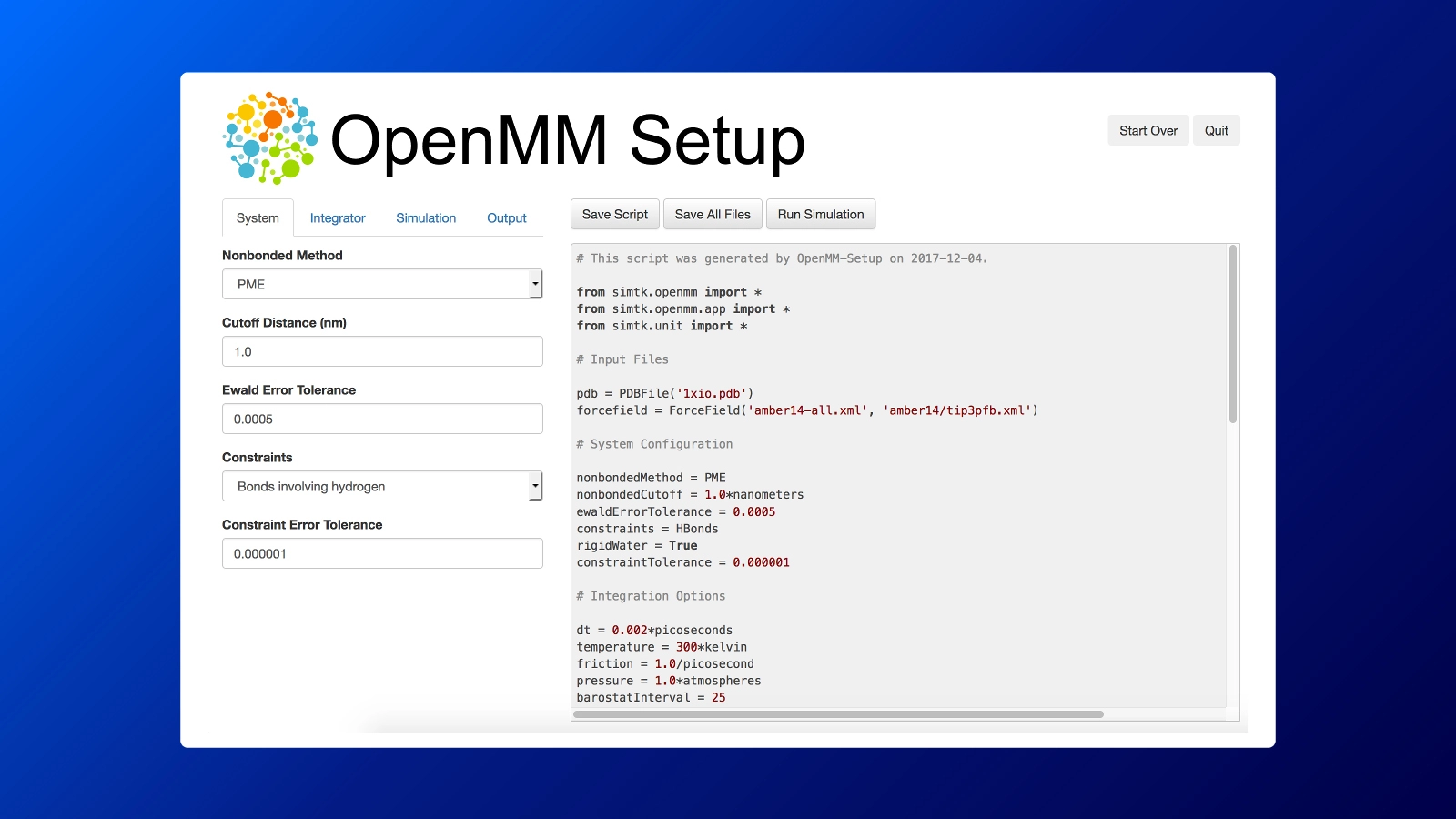
The OpenMM-Setup Application is one way to create your own scripts to start your simulations. It is more than a script builder with the ability to fix problems in input files, add missing atoms, build membranes, and much more. It is as simple as installing and opening this GUI:
#Install OpenMM-Setup
conda install -c conda-forge openmm-setup
#Launch OpenMM-Setup
openmm-setup
Since OpenMM is open source, algorithm developers can modify and edit the code to suit their needs. Users also can customize different force fields, integrators, and algorithms to support even the most niche experiments and simulations. With many different forces, angles, particles, and fields, coupled with the ability to customize them amounts to infinite possibilities in simulating various computations.
Developing your custom forces is extremely simple with OpenMM’s custom functions. Users will input their variables into a formula to create new forces for bonds, angles, torsion, and more. OpenMM also uses a simple XML file format to describe force fields. Users can use common force fields imported via XML such as Amber14 and TIP3P-FB but you can also create your own. A force field can use all the standard OpenMM force classes, as well as the very flexible custom force classes. You can even extend the Force Field class to add support for completely new forces, such as ones defined in plugins. This makes it a powerful tool for development.
Since OpenMM is built on library packages and then imported into Python to execute scripting, the ease of integration enables OpenMM to expand and develop from multiple ends. Independent developers can upload their OpenMM-supported package to extend or introduce new capabilities.
It can go both ways: the OpenMM library provides a Python interface, making it easy to integrate with other Python packages. For example, developers can use the NumPy or Pandas library to manipulate simulation data or integrate OpenMM with visualization packages such as VMD or PyMOL.
Updates to OpenMM can be modifications of the code for faster speed-ups, bug fixes, or additions to bonds, fields, and functions. However, larger more substantial additions can be worked on separately and integrated by releasing new packages.
Speaking of package integration and new capabilities, OpenMM 8.0, released February 1st, 2023, introduces the support of Machine Learning Potentials that goes beyond the core OpenMM package. Three new libraries within OpenMM enable new ways to study data using machine learning:
With a higher degree of customizability, OpenMM delivers researchers the tools to develop simulations and tests that pertain to them. Just like GROMACS, OpenMM’s open-source nature lends well to its popularity and adoption from the community.
Since OpenMM leverages the use of GPU computing resources, your large-scale simulations can benefit from the acceleration that parallelization brings to scientific computing. Exxact specializes in GPU-accelerated workstations and servers and the integration and support that OpenMM has for various GPUs make it an excellent toolkit to complement your workflow.
Looking to increase your computing for drug discovery and molecular research?
Exxact provides GPU-accelerated workstations, servers, and clusters for advancing your scientific discovery.
Contact us today for more info!
OpenMM is a high-performance molecular dynamics simulation toolkit that allows researchers to study the behavior of molecules and materials at the atomic level. It is an open-source GPU-accelerated biomolecular modeling toolkit for simulation that can be used as an application, library, or flexible programming environment.
OpenMM is open-source supported by a large community of users and developers contributing to its ongoing development and improvement. Its capabilities rival other popular Molecular Dynamics simulation suites and libraries and offers extensive programmability options and a detailed API.
Like other molecular dynamics suites, OpenMM is used to simulate a wide range of molecular systems to gain insights into their behavior and properties to design new materials, develop novel drugs, and gain a more comprehensive understanding of molecular processes.
The key defining attribute of OpenMM is its underlying architecture. OpenMM's foundation is built on C++ (with CUDA and OpenCL capabilities) for high-level computations with a Python-based application layer. Due to its easy syntax and learnability, a Python-based outward-facing interface makes scripting and running simulations extremely simple for those with little programming experience.
As a leading supplier of custom scientific workstations and servers and clusters, Exxact Corporation offers custom configurable OpenMM optimized solutions to accelerate your workflow. Talk to us today for more information!
Being open-source enables OpenMM to address many different purposes and various audiences. Chemists and biologists can utilize and tinker with the Python Application layer to run simulations through OpenMM’s simple UI. They can upload PDB files or cross-platform AMBER or GROMACS files as well. Users unfamiliar with Python can use OpenMM Setup, their Python Script Writer, making it easy to get started experimenting before having to master the more complex aspects of OpenMM.
Application developers can use the computational library to add simulation features to their programs. Since OpenMM is technically an API and library of various functions accessed through Python scripting, developers can access code within OpenMM as functions in their programs. This is especially useful when implementing molecular dynamics inside comprehensive suites: e.g. GROMACS can interface with OpenMM calculations and ACEMD now uses OpenMM as the underlying dynamics engine.
Lastly, experienced molecular dynamics engineers can develop custom algorithms with their formulas and code within OpenMM to develop new methods for novel testing. Customization is a huge plus for developing new simulation algorithms.
OpenMM’s flexibility as a toolkit, application, and library lends to its customizability. Its open-source nature enables those familiar with Python to alter the code to match their research. This also allows for ongoing development and recommendation from peers around the world. There’s a good chance that questions you may have regarding OpenMM can be answered by their forum and feedback is constantly provided to further improve its capabilities.
Developers can also utilize what OpenMM has already contributed to the Molecular Dynamics space for leveraging the expansive library to their own applications and software suites for more niche simulations. Explore their Documentation for more information.
OpenMM’s simulations can be run through a Python script with no more than 20 lines of code. They have plenty of examples of how to use OpenMM right out of the box: These scripts can look as simple as:
from openmm.app import *
from openmm import *
from openmm.unit import *
from sys import stdout
pdb = PDBFile('input.pdb')
forcefield = ForceField('amber14-all.xml', 'amber14/tip3pfb.xml')
system = forcefield.createSystem(pdb.topology, nonbondedMethod=PME,
nonbondedCutoff=1*nanometer, constraints=HBonds)
integrator = LangevinMiddleIntegrator(300*kelvin, 1/picosecond, 0.004*picoseconds)
simulation = Simulation(pdb.topology, system, integrator)
simulation.context.setPositions(pdb.positions)
simulation.minimizeEnergy()
simulation.reporters.append(PDBReporter('output.pdb', 1000))
simulation.reporters.append(StateDataReporter(stdout, 1000, step=True,
potentialEnergy=True, temperature=True))
simulation.step(10000)
But even that might seem daunting for those who haven’t touched Python. OpenMM acknowledges this and supplies a user-friendly interface with a builder that helps create fast simulations with ease.
The OpenMM-Setup Application is one way to create your own scripts to start your simulations. It is more than a script builder with the ability to fix problems in input files, add missing atoms, build membranes, and much more. It is as simple as installing and opening this GUI:
#Install OpenMM-Setup
conda install -c conda-forge openmm-setup
#Launch OpenMM-Setup
openmm-setup
Since OpenMM is open source, algorithm developers can modify and edit the code to suit their needs. Users also can customize different force fields, integrators, and algorithms to support even the most niche experiments and simulations. With many different forces, angles, particles, and fields, coupled with the ability to customize them amounts to infinite possibilities in simulating various computations.
Developing your custom forces is extremely simple with OpenMM’s custom functions. Users will input their variables into a formula to create new forces for bonds, angles, torsion, and more. OpenMM also uses a simple XML file format to describe force fields. Users can use common force fields imported via XML such as Amber14 and TIP3P-FB but you can also create your own. A force field can use all the standard OpenMM force classes, as well as the very flexible custom force classes. You can even extend the Force Field class to add support for completely new forces, such as ones defined in plugins. This makes it a powerful tool for development.
Since OpenMM is built on library packages and then imported into Python to execute scripting, the ease of integration enables OpenMM to expand and develop from multiple ends. Independent developers can upload their OpenMM-supported package to extend or introduce new capabilities.
It can go both ways: the OpenMM library provides a Python interface, making it easy to integrate with other Python packages. For example, developers can use the NumPy or Pandas library to manipulate simulation data or integrate OpenMM with visualization packages such as VMD or PyMOL.
Updates to OpenMM can be modifications of the code for faster speed-ups, bug fixes, or additions to bonds, fields, and functions. However, larger more substantial additions can be worked on separately and integrated by releasing new packages.
Speaking of package integration and new capabilities, OpenMM 8.0, released February 1st, 2023, introduces the support of Machine Learning Potentials that goes beyond the core OpenMM package. Three new libraries within OpenMM enable new ways to study data using machine learning:
With a higher degree of customizability, OpenMM delivers researchers the tools to develop simulations and tests that pertain to them. Just like GROMACS, OpenMM’s open-source nature lends well to its popularity and adoption from the community.
Since OpenMM leverages the use of GPU computing resources, your large-scale simulations can benefit from the acceleration that parallelization brings to scientific computing. Exxact specializes in GPU-accelerated workstations and servers and the integration and support that OpenMM has for various GPUs make it an excellent toolkit to complement your workflow.
Looking to increase your computing for drug discovery and molecular research?
Exxact provides GPU-accelerated workstations, servers, and clusters for advancing your scientific discovery.
Contact us today for more info!
